Dragon · Draco magnificus (Merlinus, 1022)
Abstract
We present a genome assembly from an individual male Draco magnificus (Dragon; Chordata; Reptilia; Draconiformes; Draconidae). The genome sequence has a total length of 2700 megabases. Most of the assembly (98.3%) is scaffolded into 21 chromosomal pseudomolecules, including the X and Y sex chromosomes. The mitochondrial genome assembly is 16.9 kilobases long. Gene annotation of this assembly on Ensembl identified 21500 protein-coding genes.
Background
The genome of the Dragon, Draco magnificus, was sequenced as part of the Darwin Tree of Life Project, a collaborative effort to sequence all named eukaryotic species in the Atlantic Archipelago of Britain and Ireland. Here we present a chromosomally complete genome sequence for Draco magnificus, based on a male specimen from Asia (Figure 1).
Genome sequence report
The genome of Draco magnificus (Figure 1 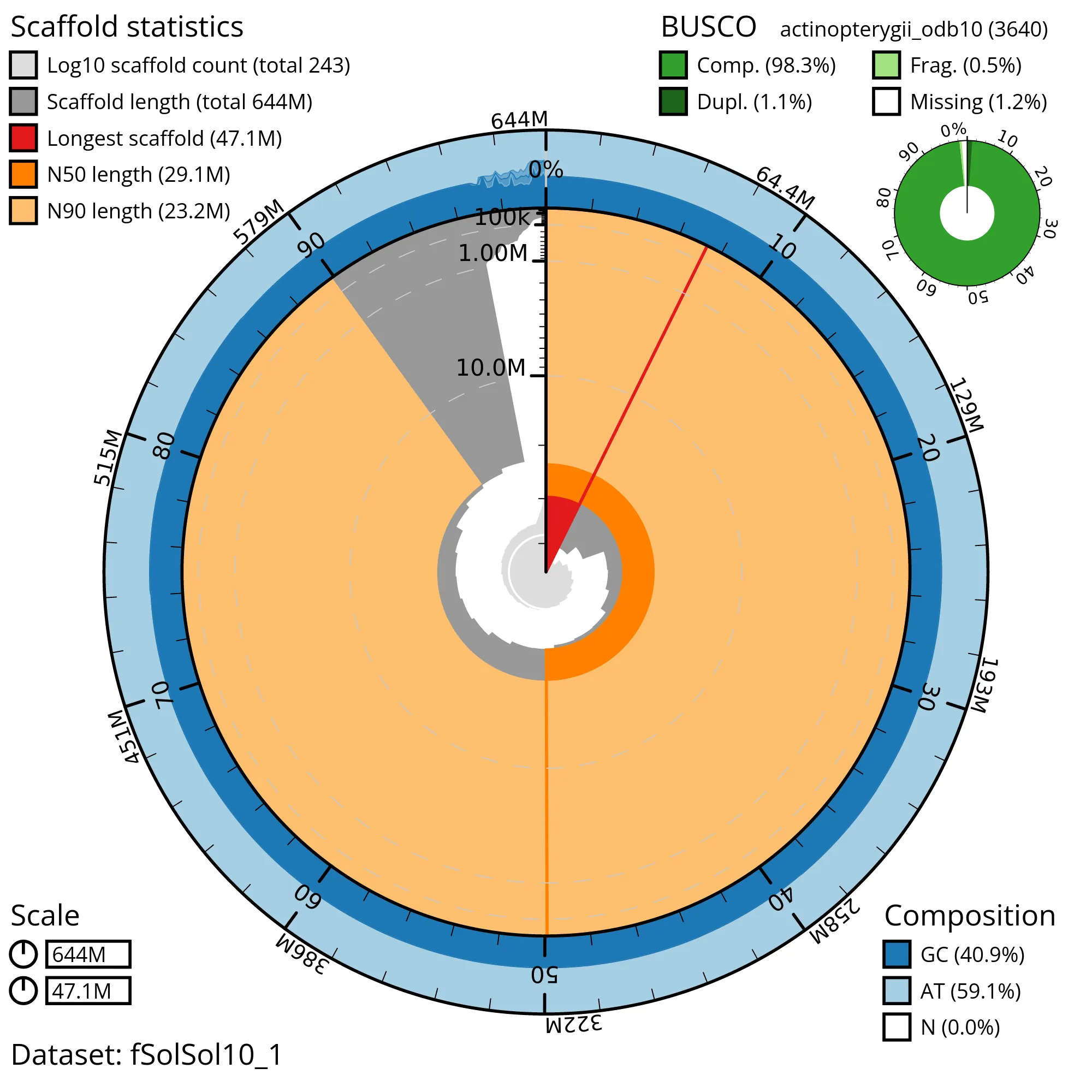 ) was sequenced using Pacific Biosciences single-molecule HiFi long reads, generating a
total of 47 Gb (gigabases) from 4.7 million
reads, providing an estimated 68-fold coverage. Primary assembly contigs
were scaffolded with chromosome conformation Hi-C data, which produced 165 Gb from 980 million reads, yielding an approximate coverage of 68-fold. Specimen and sequencing details are summarised in Table 1.
) was sequenced using Pacific Biosciences single-molecule HiFi long reads, generating a
total of 47 Gb (gigabases) from 4.7 million
reads, providing an estimated 68-fold coverage. Primary assembly contigs
were scaffolded with chromosome conformation Hi-C data, which produced 165 Gb from 980 million reads, yielding an approximate coverage of 68-fold. Specimen and sequencing details are summarised in Table 1.
Assembly errors were corrected by manual curation, including 17 missing joins or mis-joins and 3 haplotypic duplications. This reduced the assembly length by 6.3% and the scaffold number by 2%, and increased the scaffold N50 by 0.6%. The final assembly has a total length of 2700 Mb in 90 sequence scaffolds, with 260 gaps, and a scaffold N50 of 63 Mb (Table 2).
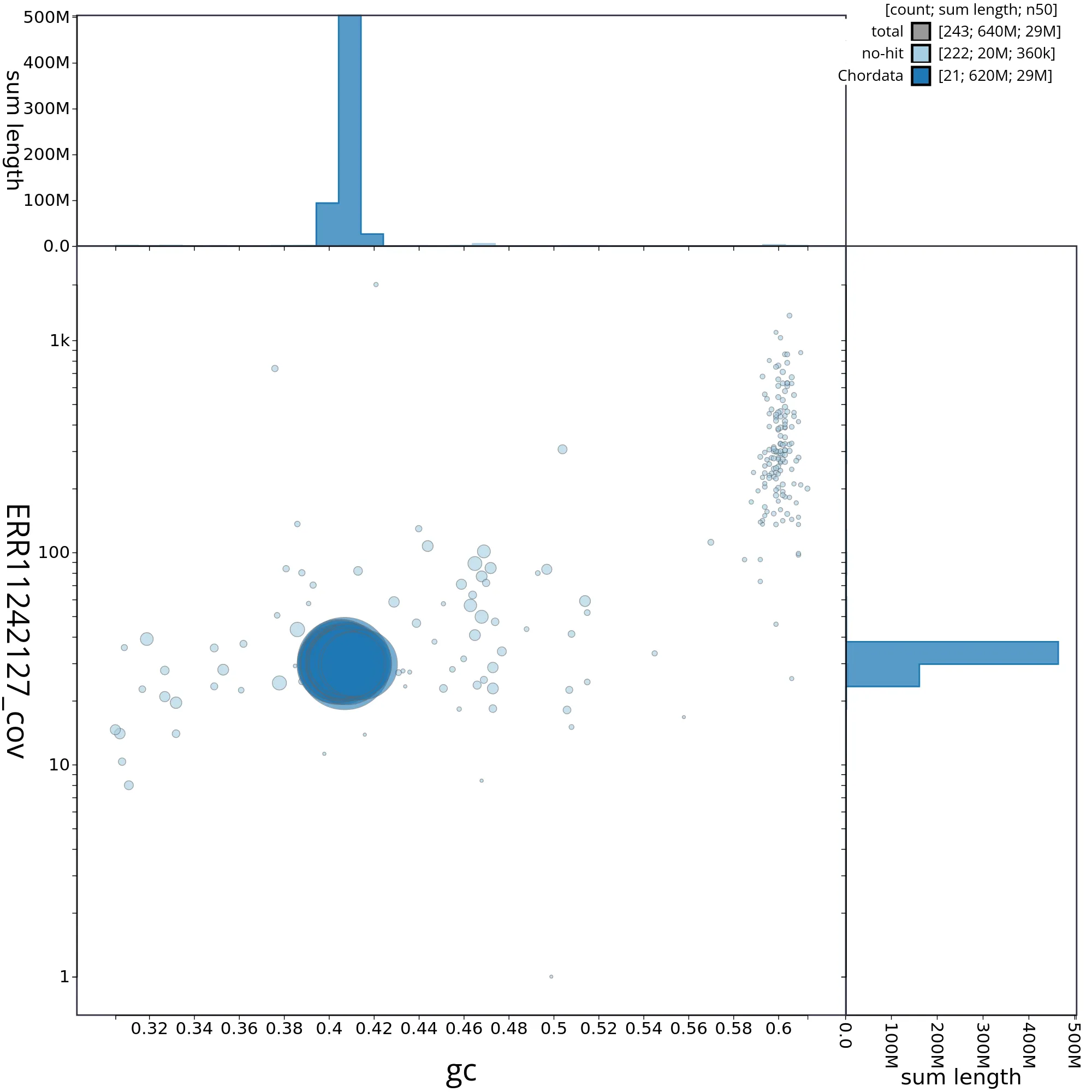
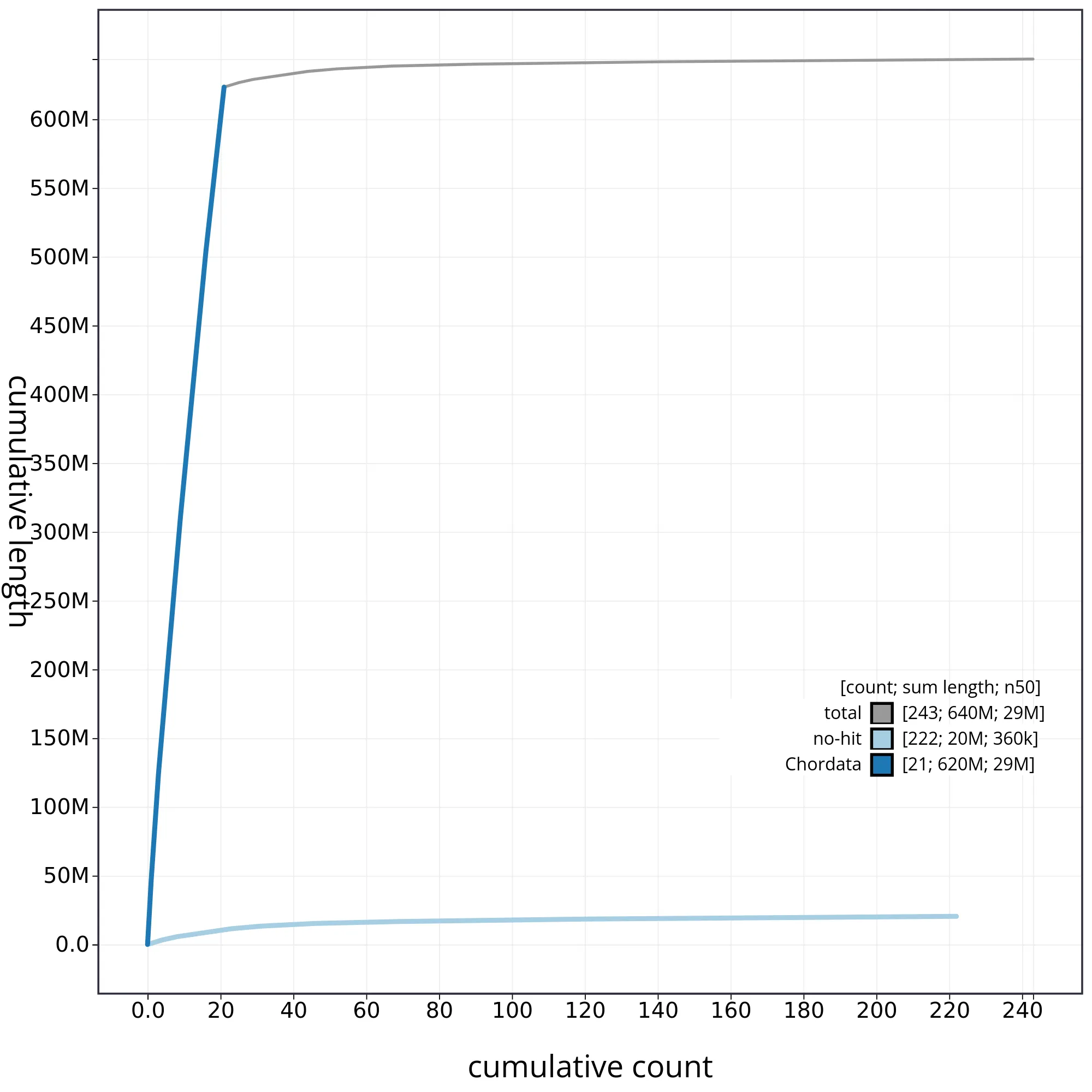
The snail plot in Figure 2 provides a summary of the assembly statistics, indicating the distribution of scaffold lengths and other assembly metrics. Figure 3 shows the distribution of scaffolds by GC proportion and coverage. Figure 4 presents a cumulative assembly plot, with separate curves representing different scaffold subsets assigned to various phyla, illustrating the completeness of the assembly.
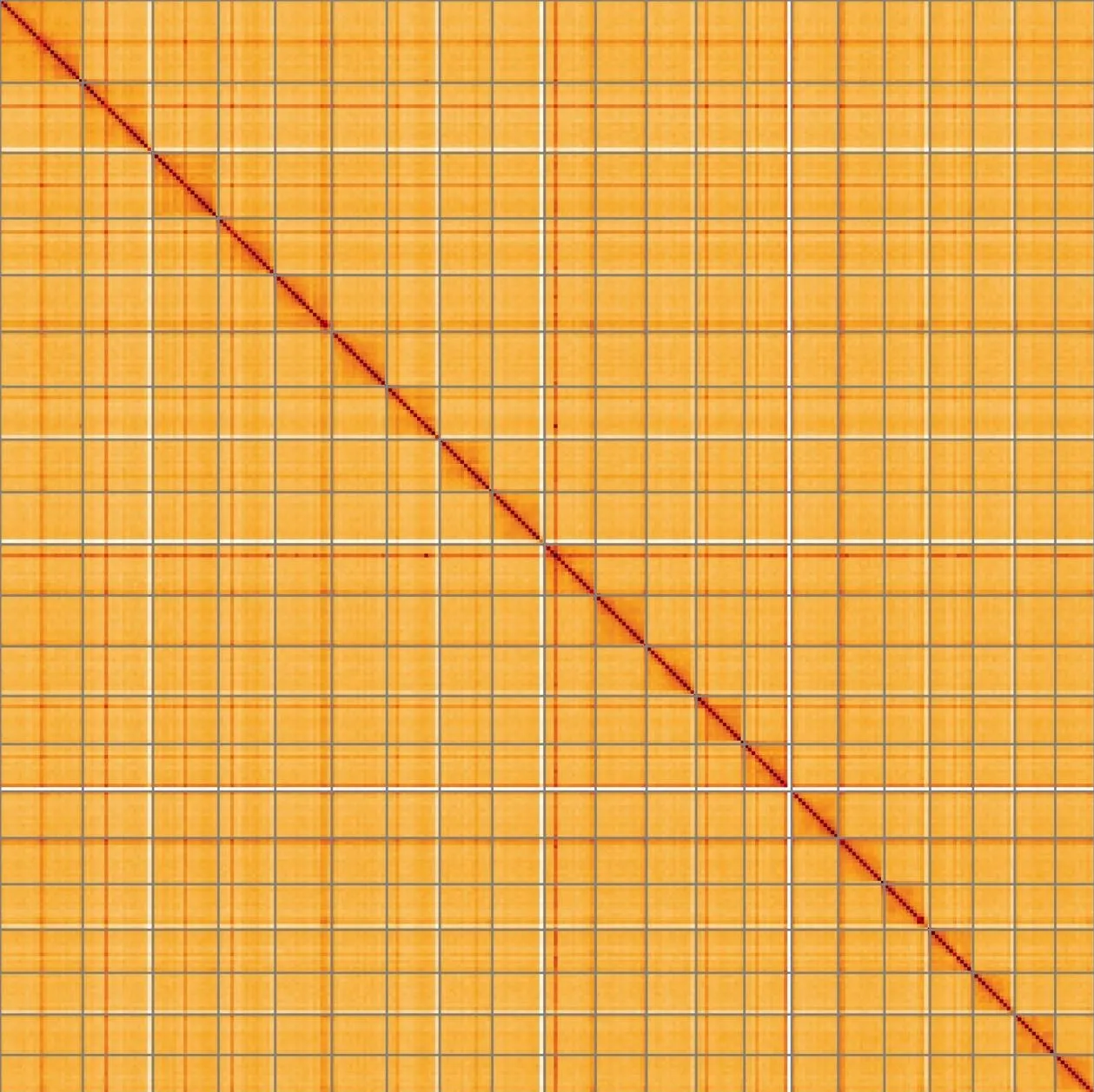
Most of the assembly sequence (98.3%) was assigned to 21 chromosomal-level scaffolds, representing 20 autosomes and the XY sex chromosome. These chromosome-level scaffolds, confirmed by the Hi-C data, are named in order of size (Figure 5; Table 3). During manual curation it was noted that This is a manual note..
The mitochondrial genome was also assembled and can be found as a contig within the multifasta file of the genome submission, and as a separate fasta file with accession GCA_890123456.1.
The final assembly has a Quality Value (QV) of 60.7 and k-mer completeness of 100%. BUSCO (v5.4.3) analysis using the Eukaryota_odb10 reference set (n = 4) indicated a completeness score of 96.2% (single = 92.2%, duplicated = 4%).
Metadata for specimens, BOLD barcode results, spectra estimates, sequencing runs, contaminants and pre-curation assembly statistics are given at https://links.tol.sanger.ac.uk/species/999002.
Methods
This genome note uses linked protocols.
| Sample preparation | |
| Sample homogenisation | Crowley et al., 2023 |
| DNA extraction, fragmentation and purification | |
| RNA extraction | |
| Hi-C preparation | Twyford et al., 2024 |
| Library preparation | |
| Sequencing | Narváez-Gómez et al., 2023 |
Sample acquisition and DNA barcoding
A male Draco magnificus (ToLID rRattus1) was collected from Asia.
The initial identification was verified by an additional DNA barcoding process according to the framework developed by Twyford et al. (2024). A small sample was dissected from the specimens and stored in ethanol, while the remaining parts were shipped on dry ice to the Wellcome Sanger Institute (WSI). The tissue was lysed, the COI marker region was amplified by PCR, and amplicons were sequenced and compared to the BOLD database, confirming the species identification (Crowley et al., 2023). Following whole genome sequence generation, the relevant DNA barcode region was also used alongside the initial barcoding data for sample tracking at the WSI (Twyford et al., 2024). The standard operating procedures for Darwin Tree of Life barcoding have been deposited on protocols.io (Beasley et al., 2023).
Nucleic acid extraction
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) Tree of Life Core Laboratory includes a sequence of core procedures: sample preparation and homogenisation, DNA extraction, fragmentation and purification. Detailed protocols are available on protocols.io (Denton et al., 2023b).
The rRattus1 sample was prepared for DNA extraction by weighing and dissecting it on dry ice (Jay et al., 2023).
Tissue from the tail was cryogenically disrupted using the Covaris cryoPREP® Automated Dry Pulverizer (Narváez-Gómez et al., 2023).
Hi-C preparation
brain tissue of the rRattus2 sample was processed at the WSI Scientific Operations core, using the Arima-HiC v2 kit. Tissue (stored at –80 °C) was fixed, and the DNA crosslinked using a TC buffer with 22% formaldehyde. After crosslinking, the tissue was homogenised using the Diagnocine Power Masher-II and BioMasher-II tubes and pestles. Following the kit manufacturer's instructions, crosslinked DNA was digested using a restriction enzyme master mix. The 5'-overhangs were then filled in and labelled with biotinylated nucleotides and proximally ligated. An overnight incubation was carried out for enzymes to digest remaining proteins and for crosslinks to reverse. A clean up was performed with SPRIselect beads prior to library preparation.
Library preparation and sequencing
Library preparation and sequencing were performed at the WSI Scientific Operations core. Pacific Biosciences HiFi circular consensus DNA sequencing libraries were prepared using the PacBio Express Template Preparation Kit v2.0 (Pacific Biosciences, California, USA) as per the manufacturer's instructions. The kit includes the reagents required for removal of single-strand overhangs, DNA damage repair, end repair/A-tailing, adapter ligation, and nuclease treatment. Library preparation also included a library purification step using AMPure PB beads (Pacific Biosciences, California, USA) and size selection step to remove templates shorter than 3 kb using AMPure PB modified SPRI. DNA concentration was quantified using the Qubit Fluorometer v2.0 and Qubit HS Assay Kit and the final library fragment size analysis was carried out using the Agilent Femto Pulse Automated Pulsed Field CE Instrument and gDNA 165kb gDNA and 55kb BAC analysis kit. Samples were sequenced using the Sequel IIe system (Pacific Biosciences, California, USA). The concentration of the library loaded onto the Sequel IIe was between 40–135 pM. The SMRT link software, a PacBio web-based end-to-end workflow manager, was used to set-up and monitor the run, as well as perform primary and secondary analysis of the data upon completion.
References
Abdennur, N. and Mirny, L. A. (2020) Cooler: Scalable storage for Hi-C data and other genomically labeled arrays, Bioinformatics, 36 (1), pp. 311–316. DOI:10.1093/bioinformatics/btz540.
Aken, B. L., Ayling, S., Barrell, D., Clarke, L., Curwen, V., Fairley, S., et al. (2016) The Ensembl gene annotation system., Database: The Journal of Biological Databases and Curation, 2016, pp. baw093. DOI:10.1093/database/baw093.
Allio, R., Schomaker‐Bastos, A., Romiguier, J., Prosdocimi, F., Nabholz, B. and Delsuc, F. (2020) MitoFinder: Efficient automated large‐scale extraction of mitogenomic data in target enrichment phylogenomics, Molecular Ecology Resources, 20 (4), pp. 892–905. DOI:10.1111/1755-0998.13160.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990) Basic local alignment search tool, Journal of Molecular Biology, 215 (3), pp. 403–410. DOI:10.1016/S0022-2836(05)80360-2.
Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Ahmad, S., Alpi, E., et al. (2023) UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Research, 51 (D1), pp. D523–D531. DOI:10.1093/nar/gkac1052.
Bates, A., Clayton-Lucey, I. and Howard, C. (2023) Sanger Tree of Life HMW DNA Fragmentation: Diagenode Megaruptor®3 for LI PacBio, protocols.io. DOI:10.17504/protocols.io.81wgbxzq3lpk/v1.
Beasley, J., Uhl, R., Forrest, L. L., Bell, D., Hart, M., Yahr, R. and Kilias, E. (2023) DNA barcoding SOPs for the Darwin Tree of Life Project, protocols.io. Available from: https://dx.doi.org/10.17504/protocols.io.261ged91jv47/v1 [Accessed 25 June 2024].
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. and Borodovsky, M. (2021) BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database, NAR Genomics and Bioinformatics, 3 (1), pp. 1–11. DOI:10.1093/nargab/lqaa108.
Buchfink, B., Reuter, K. and Drost, H.-G. (2021) Sensitive protein alignments at tree-of-life scale using DIAMOND, Nature Methods, 18 (4), pp. 366–368. DOI:10.1038/s41592-021-01101-x.
Challis, R., Kumar, S., Sotero-Caio, C., Brown, M. and Blaxter, M. (2023) Genomes on a Tree (GoaT): A versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic tree of life, Wellcome Open Research, 8, pp. 24. DOI:10.12688/wellcomeopenres.18658.1.
Challis, R., Richards, E., Rajan, J., Cochrane, G. and Blaxter, M. (2020) BlobToolKit – interactive quality assessment of genome assemblies, G3: Genes, Genomes, Genetics, 10 (4), pp. 1361–1374. DOI:10.1534/g3.119.400908.
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. and Li, H. (2021) Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm, Nature Methods, 18 (2), pp. 170–175. DOI:10.1038/s41592-020-01056-5.
Crowley, L., Allen, H., Barnes, I., Boyes, D., Broad, G. R., Fletcher, C., et al. (2023) A sampling strategy for genome sequencing the British terrestrial arthropod fauna., Wellcome Open Research, 8, pp. 123. DOI:10.12688/wellcomeopenres.18925.1.
da Veiga Leprevost, F., Grüning, B. A., Alves Aflitos, S., Röst, H. L., Uszkoreit, J., Barsnes, H., et al. (2017) BioContainers: an open-source and community-driven framework for software standardization, Bioinformatics, 33 (16), pp. 2580–2582. DOI:10.1093/bioinformatics/btx192.
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021) Twelve years of SAMtools and BCFtools, GigaScience, 10 (2). DOI:10.1093/gigascience/giab008.
Rhie, A., Walenz, B. P., Koren, S. and Phillippy, A. M. (2020) Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies, Genome Biology, 21 (1). DOI:10.1186/s13059-020-02134-9.
Sayers, E. W., Cavanaugh, M., Clark, K., Pruitt, K. D., Sherry, S. T., Yankie, L. and Karsch-Mizrachi, I. (2024) GenBank 2024 Update, Nucleic Acids Research, 52 (D1), pp. D134–D137. DOI:10.1093/nar/gkad903.
Sheerin, E., Sampaio, F., Oatley, G., Todorovic, M., Strickland, M., do Amaral, R. J. V. and Howard, C. (2023) Sanger Tree of Life HMW DNA Extraction: Automated MagAttract v.1, protocols.io. DOI:10.17504/protocols.io.x54v9p2z1g3e/v1.
Strickland, M., Cornwell, C. and Howard, C. (2023a) Sanger Tree of Life Fragmented DNA clean up: Manual SPRI, protocols.io. DOI:10.17504/protocols.io.kxygx3y1dg8j/v1.
Strickland, M., Moll, R., Cornwell, C., Smith, M. and Howard, C. (2023b) Sanger Tree of Life HMW DNA Extraction: Manual MagAttract, protocols.io. DOI:10.17504/protocols.io.6qpvr33novmk/v1.
Surana, P., Muffato, M. and Qi, G. (2023a) sanger-tol/readmapping: sanger-tol/readmapping v1.1.0 - Hebridean Black (1.1.0). Zenodo. DOI:10.5281/zenodo.7755669.
Surana, P., Muffato, M. and Sadasivan Baby, C. (2023b) sanger-tol/genomenote (v1.0.dev). Zenodo. DOI:10.5281/zenodo.6785935.
Todorovic, M., Sampaio, F. and Howard, C. (2023) Sanger Tree of Life HMW DNA Fragmentation: Diagenode Megaruptor®3 for PacBio HiFi, protocols.io. DOI:10.17504/protocols.io.8epv5x2zjg1b/v1.
Twyford, A. D., Beasley, J., Barnes, I., Allen, H., Azzopardi, F., Bell, D., et al. (2024) A DNA barcoding framework for taxonomic verification in the Darwin Tree of Life Project, Wellcome Open Research, 9, pp. 339. DOI:10.12688/wellcomeopenres.21143.1.
Figures
Reviews
All genome notes undergo automated technical reviews, with manual reviews added as needed.
Images and Figures

Assertions
Statements made by the contributing team.

